

Amyloidoza ATTR
Wszystko, co warto wiedzieć o Amyloidozie ATTR
Amyloidoza ATTR – kompendium wiedzy
Dziedziczna amyloidoza transtyretynowa (ATTR) jest bardzo rzadką chorobą nerwowo-mięśniową. Jest to choroba uwarunkowana genetycznie, w przeciwieństwie do znacznie częściej występującej tzw. dzikiej postaci amyloidozy transtyretynowej.
Mutacja w genie TTR – przyczyna dziedzicznej amyloidozy transtyretynowej
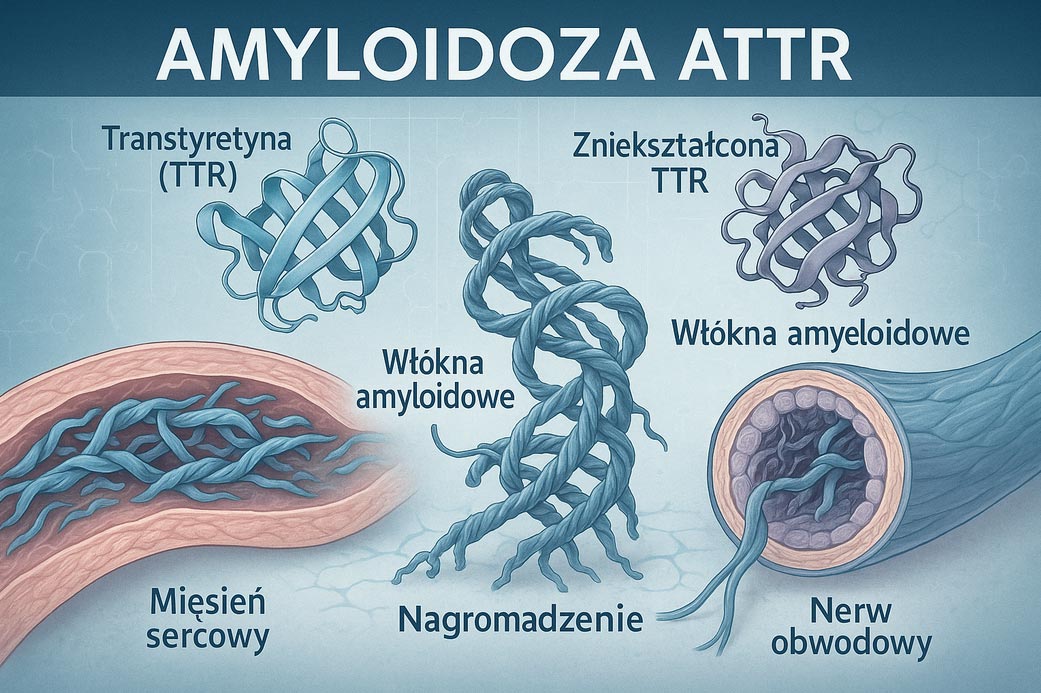
Przyczyną dziedzicznej amyloidozy transtyretynowej (ATTR) są mutacje w genie TTR, który odpowiada za produkcję w wątrobie białka – transtyretyny. U osoby zdrowej białko to służy do transportowania hormonów tarczycy i retinolu. Łączy się z nimi, przedostaje się do krwiobiegu i transportuje je – wraz z krwią – do różnych narządów.
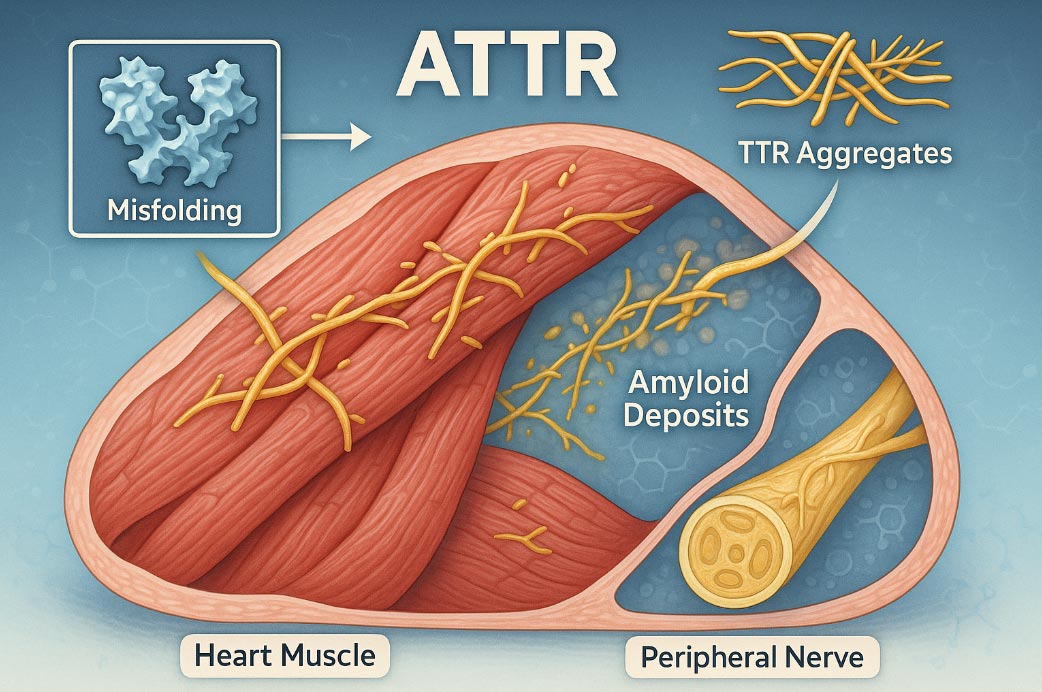
U osoby chorującej na amyloidozę transtyretynową, w wyniku mutacji w genie TTR, w wątrobie produkowana jest nieprawidłowa transtyretyna. Taka transtyretyna jest bardzo nietrwała, rozpada się, a z produktów jej rozpadu tworzą się toksyczne nitkowate struktury – włókna amyloidowe, które oklejają różne narządy. Rezultatem nagromadzenia złogów amyloidów są nieodwracalne uszkodzenia tych narządów, przede wszystkim serca i nerwów obwodowych (czyli nerwów, które docierają praktycznie do wszystkich komórek ciała). Złogi amyloidowe mogą tworzyć się również w nerkach i w gałkach ocznych, powodując występowanie różnych objawów.
Warto wiedzieć, że nie każda osoba z mutacją w genie TTR zachoruje na amyloidozę transtyretynową. Może być tak, że ktoś, kto posiada taką mutację, przeżyje całe życie bez objawów choroby. Oznacza to, że amyloidoza transtyretynowa może zostać odziedziczona po rodzicach, nawet jeśli u żadnego z nich nie występowały objawy. Jak dotąd nie ustalono, dlaczego niektóre osoby z mutacją w genie TTR zachorowują na amyloidozę, a inne nie.
Dziedziczna amyloidoza transtyretynowa najczęściej ujawnia się po 30 roku życia lub później. Typowy wiek zachorowania to 50-60 lat. W Polsce choruje na nią kilkadziesiąt osób, choć nosicieli mutacji w genie TTR jest więcej.
Zróżnicowane objawy dziedzicznej amyloidozy transtyretynowej
Objawy dziedzicznej amyloidozy transtyretynowej (ATTR) mogą się znacznie różnić u poszczególnych osób, ale zazwyczaj obejmują problemy związane z układem nerwowym i sercem. Pacjenci mogą odczuwać:
- drętwienie i mrowienie w dłoniach i stopach,
- utratę czucia w kończynach,
- nadwrażliwość na ból,
- osłabienie i skurcze mięśni,
- ogólne zmęczenie i słabość,
- zaburzenia rytmu serca,
- problemy z oddawaniem moczu.
U różnych osób choroba postępuje w różnym tempie, ale z biegiem czasu pojawiają się:
- trudności z chodzeniem,
- problemy trawienne, takie jak nasilone biegunki lub zaparcia,
- utrata masy ciała,
- kardiomiopatia czyli niewydolność serca.
Amyloidoza wpływa również na nerki, oczy i inne narządy.
Dziedziczna amyloidoza transtyretynowa (ATTR) to postępująca i wyniszczająca choroba – średnia długości życia nieleczonych pacjentów od momentu wystąpienia objawów wynosi ok. 5-10 lat. Dlatego bardzo ważne jest wczesne prawidłowe rozpoznanie tej choroby oraz rozpoczęcie leczenia. Niestety, ze względu na rzadkość występowania, diagnostyka amyloidozy TTR jest dużym wyzwaniem dla lekarzy. Statystyczna ścieżka diagnostyczna pacjenta z amyloidozą TTR to wizyty u minimum pięciu lekarzy różnych specjalności.
Tymczasem skuteczna i szybka diagnostyka daje szansę na wczesne rozpoczęcie procesu leczenia, co dla osób chorych oznacza przeżycie. Ponieważ choroba daje przede wszystkim objawy kardiologiczne i neurologiczne, pacjent z amyloidozą najczęściej trafia albo do kardiologa albo do neurologa. Jeśli trafi do neurologa, np. z powodu osłabienia siły mięśniowej lub różnych objawów autonomicznych, ze strony narządów wewnętrznych, to neurolog może rozpoznać polineuropatię, czyli uszkodzenie wielu nerwów obwodowych. Ale powodów polineuropatii jest ponad 100 i bardzo ważne jest to, żeby lekarz wpadł na to, że przyczyną polineuropatii u pacjenta może być rzadka choroba, jaką jest amyloidoza transtyretynowa.
Najskuteczniejszą metodą na szybką diagnozę dziedzicznej amyloidozy transtyretynowej jest dobra współpraca pomiędzy lekarzami różnych specjalności tj. kardiologami, neurologami, gastrologami, hepatologami oraz lekarzami pierwszego kontaktu.
Polineuropatia w przebiegu dziedzicznej amyloidozy transtyretynowej
Polineuropatia to uszkodzenie wielu nerwów obwodowych, czyli struktur odpowiedzialnych za unerwienie całego ciała. Zaburzenia działania nerwów obwodowych są przyczyną problemów m.in. z poruszaniem się i trawieniem u pacjentów z amyloidozą transtyretynową.
Objawy polineuropatii negatywnie wpływają na niezależność pacjentów, a ich postępujący charakter wiąże się z trudnościami w życiu codziennym oraz rosnącą potrzebą pomocy ze strony opiekunów. Znaczna część chorych nie jest w stanie wykonywać wielu prac domowych, staje się też niezdolna do pracy zawodowej, co w konsekwencji powoduje pogorszenie ich sytuacji finansowej.
Dziedziczna amyloidaza transyterynowa przebiegająca z polineuropatią jest zatem bardzo obciążająca, zarówno dla pacjentów, jak i ich opiekunów i rodziny. Osoby chore, a także ich bliscy doświadczają depresji, poczucia lęku i zmęczenia. Dlatego tak ważne jest jak najszybsze wdrożenie nowoczesnego leczenia (obecnie refundowanego w Polsce), a także poprawa dostępu do specjalistycznej rehabilitacji neurologicznej oraz do opieki psychologicznej.
Leczenie dziedzicznej amyloidozy transtyretynowej
Aktualnie w Unii Europejskiej zarejestrowanych jest kilka terapii lekowych, które skutecznie spowalniają rozwój dziedzicznej amyloidozy transtyretynowej i wydłużają czas życia pacjentów. Są to leki z o różnych mechanizmach działania: niektóre z nich stabilizują cząsteczkę transtyretyny, powodując, że nie przekształca się ona w amyloid, inne – działają jeszcze bliżej źródła choroby, wyciszając nieprawidłowy gen TTR i w ten sposób hamując syntezę cząsteczki transtyretyny.
Od 1 lipca 2024 w ramach programu lekowego B.162 „Leczenie pacjentów z kardiomiopatią” refundowany jest jeden z leków z grupy leków stabilizujących transtyretynę. Wskazaniem do zastosowania tej terapii jest kardiomiopatia w przebiegu amyloidozy transtyretynowej. Leczenie to jest prowadzone w ośrodkach kardiologicznych. Choć działa również w pewnym stopniu na polineuropatię, to dla pacjentów z dziedziczną amyloidozą, którzy mają uszkodzone zarówno serce, jak i nerwy obwodowe, może być ono niewystarczające.
Od 1 kwietnia 2025 obowiązuje nowy program lekowy B.170 „Leczenie dorosłych pacjentów z polineuropatią w I lub II stadium zaawansowania w przebiegu dziedzicznej amyloidozy transtyretynowej”. W jego ramach refundowany jest nowocześniejszy lek z grupy terapii genowych, czyli oddziałujący bezpośrednio na uszkodzony gen TTR. Leczenie to jest w stanie zatrzymać postęp polineuropatii, dlatego mamy nadzieję, że będzie jak najszybciej dostępne dla pacjentów w praktyce. Kwalifikacja do programu będzie przeprowadzana przez Zespół Koordynacyjny ds. Leczenia Rzadkich Chorób Neurologicznych, powoływany przez prezesa Narodowego Funduszu Zdrowia.
Dziedziczna amyloidoza transtyretynowa (ATTR) – ulotka w PDF
Pobierz kompendium wiedzy o dziedzicznej amyloidozie transtyretynowej (ATTR) w formie przejrzystego dokumentu PDF. Materiał w przystępny sposób wyjaśnia, czym jest choroba, jakie daje objawy, jak przebiega diagnostyka oraz jakie są możliwości leczenia i opieki nad pacjentem. To rzetelne źródło informacji dla chorych, ich rodzin i opiekunów. Dokument możesz wygodnie wydrukować lub przesłać dalej lekarzowi, bliskim lub innym osobom poszukującym sprawdzonej wiedzy o amyloidozie ATTR.
Amyloidoza transyretynowa – Ciekawostki i newsy







Amyloidoza – filmy edukacyjne dr n. med. Marty Lipowskiej z Kliniki Neurologii Warszawskiego Uniwersytetu Medycznego
Amyloidoza transtyretynowa to rzadka, postępująca choroba ogólnoustrojowa, która bardzo często zajmuje układ nerwowy. Objawy neurologiczne bywają niespecyficzne, rozwijają się stopniowo i przez długi czas mogą przypominać inne, znacznie częstsze schorzenia – co opóźnia rozpoznanie i leczenie.
W cyklu krótkich filmów edukacyjnych dr n. med. Marta Lipowska z Kliniki Neurologii Warszawskiego Uniwersytetu Medycznego – Ośrodka Eksperckiego Chorób Rzadkich Nerwowo-Mięśniowych Europejskiej Sieci Referencyjnej ERN EURO-NMD w przystępny sposób wyjaśnia, czym jest amyloidoza transtyretynowa, jakie objawy neurologiczne powinny wzbudzić czujność, jak wygląda diagnostyka oraz jakie są obecnie możliwości leczenia tej choroby.

Potrzeba opieki wielospecjalistycznej
w dziedzicznej amyloidozie transtyretynowej
Dziedziczna amyloidoza transyretynowa jest chorobą wielonarządową, dlatego leczeniem pacjentów powinien zajmować się zespół terapeutyczny, składający się ze specjalistów w różnych dziedzinach medycyny, m.in.: neurologa, kardiologa, gastrologa, rehabilitanta, niekiedy okulisty, nefrologa i psychologa. Na dodatek wszyscy ci specjaliści powinni mieć wiedzę na temat odrębności jednostki chorobowej, jaką jest amyloidoza transtyretynowa.
Bardzo potrzebne jest więc utworzenie wielospecjalistycznych ośrodków, w których pacjenci byliby objęci opieką kompleksową. Inaczej, gdy pacjent musi chodzić do każdego z lekarzy specjalistów do różnych placówek, bez żadnej koordynacji tych wizyt, jest bardzo trudno o dobrą opiekę. Dlatego w wielu krajach istnieją już centra kompleksowej diagnostyki i leczenia amyloidozy. To rozwiązanie, które znacząco poprawia jakość życia i wyniki leczenia pacjentów z amyloidozą transtyretynową. Jest ono też efektywne kosztowo z puntu widzenia systemu, ponieważ pacjent nie krąży pomiędzy ośrodkami.