

Choroba Pompego
Wszystko, co warto wiedzieć o chorobie Pompego
Choroba Pompego – kompendium wiedzy
Choroba Pompego jest spowodowana niedoborem kwaśnej alfa-glukozydazy, enzymu lizosomalnego uczestniczącego w procesie rozkładu glikogenu do glukozy. Następstwem jest nadmierne gromadzenie które prowadzi do akumulacji glikogenu szczególnie w mięśniach szkieletowych i oddechowych, mięśniu sercowym, a także w wątrobie i układzie nerwowym, powodując osłabienie mięśni i przedwczesną śmierć z powodu niewydolności oddechowej lub niewydolności serca. Objawy zależą od stopnia zachowanej resztkowej aktywności enzymu. Całkowity brak aktywności prowadzi do ciężkich zaburzeń w pierwszych miesiącach życia, obejmujących uogólnioną wiotkość i kardiomiopatię przerostową ze zwężeniem drogi odpływu z lewej komory (postać o wczesnym początku). Częściowy niedobór enzymu może się ujawnić w każdym wieku nieswoistymi objawami miopatii proksymalnej (niedowład obręczowo-kończynowy) i niewydolności oddechowej, zazwyczaj bez współistniejącej kardiomiopatii (postać o późnym początku).
Dwie postacie choroby Pompego:
Choroba Pompego w postaci niemowlęcej
W pierwszych tygodniach życia choroba powoduje ogólną wiotkość mięśni, kardiomiopatię przerostową, niewydolność oddechową, trudności w karmieniu i opóźnienie wzrastania. Objawy mogą być wykryte już w okresie prenatalnym. U wielu dzieci stwierdza się powiększenie wątroby i języka oraz niedosłuch. Przerost mięśnia sercowego prowadzi do zwężenia drogi odpływu z lewej komory oraz ucisku płuc. Mogą wystąpić zaburzenia czynności układu przewodzącego serca. Nieleczona choroba prowadzi zazwyczaj do zgonu w pierwszym roku życia. Powyżej drugiego miesiąca życia, zaburzenia są zwykle mniej nasilone. Głównym objawem klinicznym jest obniżenie napięcia mięśni i opóźnienie rozwoju ruchowego. z powodu odkładania się glikogenu mięśnie są twarde i mają konsystencję gumy. Przerost łydek i objaw Gowersa mogą sugerować dystrofię mięśniową Duchenne’a, ale objawy te występują w młodszym wieku niż w dystrofinopatii.
Choroba Pompego w postaci o późnym początku
Główną cechą jest niedowład proksymalny mięśni objawiający się trudnościami we wchodzeniu po schodach, wstawaniu z łóżka lub krzesła oraz unoszeniu ramion. Obraz kliniczny może przypominać dystrofię mięśniową obręczowo-kończynową, często współistnieje niewydolność oddechowa. Po zabiegu w znieczuleniu ogólnym wydolność oddechowa może się gwałtownie pogorszyć, powodując konieczność przewlekłego wspomagania oddychania. w wieku młodzieńczym występuje mniejsza sprawność ruchowa w porównaniu z rówieśnikami. U dzieci zauważalne może być powiększenie języka i/lub wątroby, może dojść do rozwoju hiperlordozy lędźwiowej i skoliozy. Wielu chorych, którzy zachorowali w wieku dorosłym, prowadziło wcześniej normalną aktywność życiową bez zauważalnych ograniczeń sprawności fizycznej.
Leczenie obejmuje terapię przyczynową za pomocą enzymatycznej terapii zastępczej, która polega na przewlekłej substytucji alglukozydazy alfa, czyli rekombinowanego analogu naturalnego enzymu oraz terapie objawowe poszczególnych zaburzeń, np. niewydolności serca lub niewydolności oddechowej.
Choroba może się rozwijać podstępnie przez wiele lat. Retrospektywnie dorośli pacjenci opisują często mniejszą sprawność ruchową już w wieku dziecięcym. U niektórych osób pierwszą stwierdzoną nieprawidłowością jest zwiększone stężenie transaminaz, sugerujące początkowo chorobę wątroby, a w rzeczywistości związane z uszkodzeniem mięśni, co potwierdza podwyższona aktywność kinazy kreatynowej (CK). Dostępne są testy przesiewowe suchej kropli krwi. Rozpoznanie ustala się na podstawie obniżonej aktywności kwaśnej alfa-glukozydazy oraz wyniku badania genetycznego, potwierdzającego występowanie wariantów patogennych w obu allelach genu GAA. Leczenie przyczynowe polegające na enzymatycznej terapii zastępczej zwiększa przeżywalność dzieci z postacią wczesną oraz stabilizuje lub poprawia sprawność ruchową i wydolność oddechową pacjentów z postacią późną.
Choroba Pompego – kategoryzacja medyczna
Nazwa: choroba Pompego (glikogenoza typu 2)
Nazwa angielska: Pompe disease (glycogen storage disease II – GSD II)
Częstość występowania: 1/40 000 (postać o wczesnym początku 1/140 000, postać o późnym początku 1/60 000)
Dziedziczenie: autosomalne recesywne
ICD-10: E74.0 / E74.026
MIM: 232 300
GeneReviews: NBK1261
ORPHA: 365
GARD: 5714
Gen: GAA (LRG_673)
Białko: kwaśna alfa-glukozydaza
Wiek wystąpienia choroby: może wystąpić w każdym wieku
Synonimy dla nazwy Choroba Pompego
- glikogenoza typu 2 (glycogenosis type II)
- choroba spichrzania glikogenu typu 2 (glycogen storage disease II – GSD2)
- niedobór alfa-glukozydazy lizosomalnej (lysosomal alpha-glucosidase deficiency)
- niedobór kwaśnej maltazy (acid maltase deficiency)
- niedobór kwaśnej alfa-1,4-glukozydazy (alpha1,4glucosidase deficiency)
- niedobór GAA (GAA deficiency)
Choroba Pompego – Ciekawostki i newsy




















Wywiad lekarski jest pierwszym etapem badania, które z reguły przeprowadza lekarz. Jest on ważnym elementem diagnostyki. Pomaga on w zebraniu kluczowych informacji dotyczących stanu zdrowia oraz do wysnucia wstępnych wniosków w kierunku diagnozy. W przypadku choroby Pompego, ze względu na podobieństwo objawów do innych schorzeń, lekarz powinien zlecić szereg specjalistycznych badań w celu ostatecznego potwierdzenia diagnozy
#chorobaPompego #wywiadLekarski

Przygotowanie pacjenta do wywiadu lekarskiego jest ważnym elementem, który wpływa na otrzymanie prawidłowej diagnozy. Osoba przygotowująca się do badania powinna zwrócić uwagę na charakter dolegliwości. W jakich okolicznościach one powstają. Kiedy się nasilają, a kiedy łagodzą. Podczas wizyty powinno się także posiadać listę przyjmowanych leków z dawkowaniem oraz wcześniejszą dokumentację medyczną, która dotyczy schorzenia. Ważna jest także szczerość. Zatajanie jakichkolwiek faktów dotyczących choroby może prowadzić do nieprawidłowej diagnozy lub do wydłużenia czasu jej trwania
#wywiadLekarski

Szukając informacji na temat chorób genetycznych do których należy choroba Pompego możemy spotkać się z terminem genotyp. Nazwa ta oznacza całkowity zestaw genów, który posiada organizm. Wszystkie istoty żywe, które mają materiał genetyczny, czyli bakterie, rośliny zwierzęta oraz oczywiście człowiek mają swój genotyp. Genotyp zawiera pełną informację genetyczną dotyczącą danego organizmu. Każdy osoba posiada swój unikalny genotyp, wyjątkiem są tutaj bliźnięta jednojajowe

Fenotyp można określić jako wynik oddziaływania środowiska na genotyp. Oznacza on wszystkie cechy wykształcone podczas rozwoju organizmu. Możemy do nich zaliczyć np. budowę ciała, kolor włosów, oczu, ale także skłonność do chorób lub temperament. W genotypie mamy zapisaną pełną informację genetyczną, jednak to jak rozwiną się poszczególne cechy zależy od środowiska. Przykładem może być wzrost u człowieka. Dziecko mające wysokich rodziców ma predyspozycje do takiego wzrostu. Jednak gdy nie będzie miało ono dostępu do odpowiednich składników odżywczych lub zacznie palić papierosy w okresie dojrzewania, to może nie osiągnąć genetycznie zaprogramowanej wysokości ciała. Warto zauważyć, że te same genotypy w różnych środowiskach mogą dać różne fenotypy. Możemy się spotkać także z sytuacją odwrotną, czyli różne genotypy pod wpływem środowiska mogą dać podobne fenotypy

Za choroby genetyczne w tym chorobę Pompego odpowiadają mutacje genów, czyli nieprawidłowości w ich budowie. Część z nich jest dziedziczona, a niektóre powstają samoistnie. Podczas badań w genotypie można wykryć poszczególne mutacje genów odpowiadające za dane choroby lub predyspozycje do danego schorzenia. Termin penetracja mutacji genu oznacza częstość wystąpienia choroby w populacji wszystkich nosicieli nieprawidłowego genu. W niektórych przypadkach może być ona równa 100%. Oznacza to, że u wszystkich nosicieli wystąpi schorzenie. Na zmniejszenie penetracji mogą mieć wpływ czynniki środowiskowe np. styl życia. Istnienie tego zjawiska jest niekorzystne w diagnostyce oraz przywidywaniu ryzyka wystąpienia choroby u krewnych pacjenta
#chorobyGenetyczne

Termin ekspresja genu w skrócie oznacza z jaką częstotliwością dany gen daje podobne objawy choroby. Jeżeli ekspresja genu jest zmienna to osoby chore na to samo schorzenie mogą mieć zupełnie inne objawy z różnym nasileniem. Jeżeli ekspresja genu jest stała oraz penetracja bliska 100%, to taka korelacja jest bardzo przydatna w diagnostyce danego schorzenia. Za jedną chorobę genetyczną mogą odpowiadać różne mutacje danego genu. Na przykład w chorobie Pompego wyodrębniono ponad 100 mutacji genu GAA, które są za nią odpowiedzialne. Poszczególne mutacje odpowiadające za jedno schorzenie mogą mieć też różną ekspresję genu
#chorobyGenetyczne

Ważną rolę w chorobie Pompego odgrywa badanie nieprawidłowości genu odpowiedzialnego za to schorzenie. W Centrum Medycznym Uniwersytetu Erasmusa w Roterdamie zostało zidentyfikowane i skatalogowane ponad 100 mutacji i liczne warianty genu GAA odpowiedzialnego za chorobę Pompego. Zauważono, że istnieją pewne typowe mutacje z przydatnymi korelacjami genotyp-fenotyp podczas diagnostyki. Są także mniej specyficzne i wiele rzadkich, które są w tym wypadku mniej przydatne. Badanie mutacji odgrywa ważną rolę w identyfikowaniu nosicieli w rodzinie, w której ona wystąpiła. Na podstawie wiedzy zebranej w bazach danych na całym świecie, można także oszacować procentowe występowanie danych mutacji w poszczególnych populacjach. Może to wpływać korzystnie na poprawę diagnostyki na danym terenie
#chorobaPompego

Jednym z najbardziej zajętych organów przez chorobę Pompego o późnym początku jest przepona. Jej dysfunkcja prowadzi do niewydolności oddechowej. W związku z tym ocena czynności przepony może być ważna podczas monitorowania progresji choroby oraz skuteczności leczenia. Grupa włoskich lekarzy przeprowadziła badania i wykazała, że do oceny funkcji przepony wystarczające może być jedynie badanie USG przy ustaleniu pewnych założeń. Udowodniono, że ocena uzyskana w ten sposób istotnie koreluje z wynikami badania spirometrycznego. Przewagą takiego badania nad spirometrią jest:
- większa dostępność
- mniejszy koszt
- mniejsze zaangażowanie pacjenta przy przeprowadzaniu badania
#chorobaPompego

1 stycznia 2025 roku Polska obejmie prezydencję na okres 6 miesięcy w Radzie Unii Europejskiej. Podczas trwania prezydencji kraj nasz będzie przewodniczyć posiedzeniom Rady na wszystkich szczeblach, w tym dotyczącym zdrowia. Każdy kraj wcześniej przygotowuje program na czas prezydencji oraz wyznacza priorytety. W październiku tego roku odbył się okrągły stół pt. „HEALTHCARE POLICY SUMMIT: PRIORYTETY ZDROWOTNE POLSKIEJ PREZYDENCJI W RADZIE UNII EUROPEJSKIEJ 2025” mający na celu omówienie zagadnień dotyczących zdrowia. Między innymi poruszona została kwestia chorób rzadkich. Zauważono, że Polska może stanowić dobry przykład dla innych krajów w zakresie leczenia oraz diagnozowania takich chorób. Jednym z priorytetów wpływających na optymalizację leczenia są nowoczesne techniki diagnostyczne oraz realne wzmocnienie istniejących ośrodków eksperckich. Podkreślono, że istnieje problem z organizacją opieki zdrowotnej nad dorosłymi pacjentami z chorobami rzadkimi nie tylko w Polsce, ale także na poziomie europejskim. Stwierdzono, że jednym z celów prezydencji Polski powinna być harmonizacja programów badań przesiewowych na poziomie europejskim
#chorobyRzadkie

W chorobie Pompego na skutek mutacji genu w tkance mięśniowej akumulowany jest glikogen. Skutkuje to jej uszkodzeniem. Mięśnie to nie jedyne miejsce, gdzie jest on odkładany. Magazynowany jest także w innych obszarach organizmu, w tym w mózgu. W związku z tym zakres zniszczeń występujących w organizmie chorego jest dużo większy niż przypuszczano w początkowych fazach badań choroby
#chorobaPompego

U niektórych pacjentów z dziecięcą chorobą Pompego odnotowano utratę słuchu. Może być ona spowodowana zajęciem aparatu przewodzącego lub czasami ślimaka. Pacjenci ze zdiagnozowaną chorobą Pompego oprócz standardowych badań przesiewowych słuchu u noworodków powinni mieć wykonywane, co roku podstawowe, rutynowe badania w tym kierunku. Ważną kwestią jest także ostrożna interpretacja badań przeprowadzonych przez audiologów. Dzieci, które chorują na to schorzenie stanowią wyzwanie dla lekarzy. Przy tym badaniu powinno brać się pod uwagę ich opóźnienia motoryczne oraz zwiększoną częstość występowania zajęcia ucha środkowego. Przy wykryciu nieprawidłowości należy podjąć leczenie, włącznie z interwencją chirurgiczną
#chorobaPompego

W chorobie Pompego mogą wystąpić zmiany w układzie krwionośnym. Zanotowano przypadki magazynowania glikogenu w naczyniowych mięśniach gładkich tętnic: szyjnej wewnętrznej, podstawnej i mózgowych przyśrodkowych. Są to bardzo groźne zmiany, które mogą powodować tętniaki i pęknięcia naczyń krwionośnych. Skutkować to może śmiercią chorego. Dlatego podczas badania pacjentów ważne jest, aby wziąć pod uwagę możliwość zajęcia takich naczyń krwionośnych. U niektórych pacjentów zanotowano także gorączkę o nieznanej etiologii. Istnieją przypuszczenia, że może być ona pochodzenia centralnego.
#chorobaPompego

Choroba Pompego jest jedną z nielicznych chorób rzadkich, dla których opracowano skuteczną terapię. Leczenie odbywa się po kwalifikacji do programu lekowego B.22. Jednak to nie jedyne leczenie, które powinien otrzymać chory. Biorąc pod uwagę, że choroba Pompego może atakować bardzo wiele narządów, to pacjentem powinien opiekować się zespół lekarzy różnych specjalizacji. Zajmują się oni łagodzeniem poszczególnych objawów. Mogą być to lekarze specjaliści z dziedzin:
- interny
- neurologii
- ortopedii
- kardiologii
- fizjoterapii
- dietetyki
- pulmonologii
- gastroenterologii
Obecna wiedza medyczna nie pozwala na całkowite wyleczenie choroby, jednak dostępne terapie w znacznym stopniu łagodzą objawy i hamują rozwój zaburzeń
#chorobaPompego

Programy lekowe dotyczą wybranych jednostek chorobowych i ściśle określonych pacjentów. Leczenie jest bezpłatne i odbywa się przy zastosowaniu innowacyjnych środków leczniczych, które nie są finansowane w ramach innych świadczeń gwarantowanych. Treść każdego programu lekowego jest publikowana jako załącznik do obwieszczenia Ministra Zdrowia w sprawie leków refundowanych. Zawarte są tam między innymi kryteria kwalifikacji , kryteria wyłączenia z programu, wykaz badań diagnostycznych. Decyzję o kwalifikacji podejmuje lekarz placówki mającej kontrakt w danym zakresie, na podstawie kryteriów włączenia do programu i przeprowadzonych badań
#programyLekowe

AOTMiT zatwierdził nowy lek na chorobę Pompego.
Nowy lek o nazwie Nexviadyme (awalglukozydaza alfa) został zatwierdzony przez Agencję Oceny Technologii Medycznych i Taryfikacji.
Program: B.22 – LECZENIE PACJENTÓW Z CHOROBĄ POMPEGO (ICD-10: E74.0)
Doprecyzowanie: Leczenie chorych na chorobę Pompego z udokumentowanym brakiem lub głębokim niedoborem alfa-glukozydazy.
Druga opcja terapeutyczna stanowiąca enzymatyczną terapię zastępczą obok alglukozydazy alfa. (Choroba rzadka).
Lek będzie refundowany.
Więcej informacji i źródło:
https://politykazdrowotna.com/artykul/lista-lekow-refundowanych/1206343
Artykuły o chorobie Pompego
Choroby rzadkie (ang. rare diseases)
Choroby rzadkie to te jednostki chorobowe, które w danej populacji występują u mniej niż 5 na 10 000 jej członków. Są to schorzenia zwykle o przewlekłym i ciężkim przebiegu, najczęściej uwarunkowane genetycznie, w połowie przypadków ujawniające się w wieku dziecięcym. Liczba opisanych dotychczas chorób rzadkich jest duża – wynosi ok. 6 tysięcy, co powoduje, że choruje na nie łącznie ok. 6-8% populacji. W Polsce to ok. 2,3-3mln osób. Dokładne rozpoznanie tych chorób nie jest zawsze możliwe, ponieważ większość lekarzy nie miała szansy się z nimi wcześniej spotkać ze względu na ich rzadkie występowanie. W związku z tym droga do ustalenia diagnozy dla wielu pacjentów jest długa. W miarę doskonalenia metod diagnostycznych, a przede wszystkim dostępności nowoczesnych terapii, wiedza na temat chorób rzadkich stale się poszerza.
Choroby lizosomalne spichrzeniowe
To inaczej choroby spichrzania lizosomalnego, jest to grupa kilkudziesięciu chorób uwarunkowanych genetycznie. Choroby spichrzeniowe dzielimy na mukopolisacharydozy (MPS), glikoproteinozy (np. sjalidoza, choroba Schindlera), sfingolipidozy, w tym cerebrozydozy (np. choroba Fabry’ego, Farbera, Gauchera, Krabbego, Niemanna-Picka), glikogenozy (choroba Pompego), lipidozy (choroba Wolmana), zaburzenia transportu lizosomalnego (np. cystynoza, choroba spichrzania kwasu sjalowego – choroba z Salli) bądź mnogie niedobory enzymatyczne (niedobór sulfataz, mukolipidoza). U podłoża których leży defekt enzymatyczny (głównie hydrolaz, rzadziej peptydaz). W wyniku zaburzeń aktywności różnych enzymów dochodzi do odkładania/akumulacji/gromadzenia niechcianych substancji wewnątrz organelli komórkowych, głównie w lizosomach. Choroby te biorą swoje nazwy od konkretnych substratów, których metabolizm jest w danym przypadku zaburzony, co skutkuje nagromadzeniem w komórkach – mukopolisacharydów, lipidów, sulfatydów, sfingolipidów, gangliozydów, glikoprotein itd. Prowadzi to do określonych zmian histochemicznych, strukturalnych i czynnościowych w organizmie wskutek powiększenia komórek i w konsekwencji całych narządów. Pierwszą opisaną chorobą lizosomalną była choroba Pompego. W Polsce częstość chorób lizosomalnych spichrzeniowych szacuje się na 1/23 tys. żywych urodzeń. Dziedziczenie tych chorób jest najczęściej autosomalne recesywne, jedynie choroba Fabry’ego, Huntera i Danona dziedziczą się w sposób dominujący sprzężony z płcią. Podstawą leczenia chorób lizosomalnych spichrzeniowych jest enzymatyczna terapia zastępcza odwracająca niedomogę danego enzymu – stosowana np. w chorobie Gauchera czy chorobie Fabry’ego. Wykonuje się również przeszczepienie szpiku kostnego, np. w MPS i leukodystrofii metachromatycznej. Metody nieenzymatyczne obejmują lecznicze zmniejszenie produkcji substratu, np. w niedoborach gluko- czy galaktozydaz. Niektóre choroby spichrzeniowe prowadzą do zgonu już w niemowlęctwie. Inne zaczynają się w wieku młodzieńczym lub dorosłym.
Objawy kliniczne choroby Pompego
Nieprawidłowa aktywność enzymów/koenzymów prowadzi do postępującego osłabienia mięśni – najwyraźniej proksymalnych, trudności z chodzeniem, wchodzeniem po schodach, arefleksji, duszności wysiłkowej, zmęczenia, trudności w karmieniu w okresie niemowlęcym, zwiększonego wydalania z moczem oligosacharydów pochodzących z częściowej degradacji glikoprotein, kardiomegalii, hepatomegalii, nieprawidłowości miopatycznych w badaniu EMG, podwyższonej aminotransferazy alaninowej, kinazy kreatynowej, dehydrogenazy mleczanowej w surowicy, akumulacji glikogenu w lizosomach włókien mięśniowych, objaw Gowersa, szmerów serca oraz infekcje dróg oddechowych. Wspólną cechą kliniczną pacjentów z chorobą Pompego jest postępujące nieswoiste osłabienie mięśni. Początek kliniczny, stopień niedowładu i szybkość postępowania mieszczą się jednak w bardzo szerokich granicach w zależności od zachowanej resztkowej aktywności kwaśnej alfa-glukozydazy. U niemowląt <12. mies. życia z całkowitym niedoborem lub śladową aktywnością enzymu (<1% wartości referencyjnych), występuje postać o wczesnym początku, cechująca się uogólnioną wiotkością, kardiomiopatią przerostową i szybkim postępowaniem objawów. U pacjentów z częściowym niedoborem enzymu (2–40% wartości referencyjnych) rozwija się postać o późnym początku bez kardiomiopatii. Może się ona ujawnić klinicznie w każdym wieku >12. miesiąca życia, również u dorosłych, nawet >60 lat. Zalicza się do niej także przypadki o początku <12. miesiąca życia bez istotnego przerostu mięśnia sercowego. Postać o wczesnym początku odpowiada za nie więcej niż 20% przypadków. Częstość występowania postaci o późnym początku jest prawdopodobnie niedoszacowana. Ze względu na mało swoiste objawy u wielu chorych może pozostawać nierozpoznana. W wieku wczesnodziecięcym uwagę zwraca najczęściej opóźnienie rozwoju ruchowego, a u młodzieży i dorosłych postępujący niedowład proksymalny (obręczowo-kończynowy). Stosunkowo duże osłabienie mięśni tułowia, w tym mięśni oddechowych, prowadzi do wczesnej niewydolności oddechowej (wyprzedzającej zazwyczaj utratę zdolności samodzielnego chodu).
Badania diagnostyczne w chorobie Pompego
Pomimo rzadkiego występowania chorobę Pompego należy uwzględnić na wczesnym etapie diagnostyki różnicowej miopatii ze względu na możliwość skutecznego leczenia przyczynowego (enzymatyczna terapia zastępcza).
Różnicowanie choroby Pompego
Diagnostyka różnicowa zależy od wieku: w okresie noworodkowym i niemowlęcym obejmuje inne przyczyny zespołu dziecka wiotkiego (zwłaszcza współistniejące z powiększeniem mięśnia sercowego), a w późniejszym okresie życia – dystrofinopatie, różne postacie dystrofii obręczowo-kończynowej i miopatie nabyte.
Diagnostyka różnicowa choroby Pompego – postać o wczesnym początku:
- rdzeniowy zanik mięśni
- glikogenoza typu IIIa
- glikogenoza typu IV
- idiopatyczna kardiomiopatia przerostowa
- zespół Danona
- zwłóknienie sprężyste wsierdzia (fibroelastoza wsierdzia)
- zapalenie wsierdzia
- choroby mitochondrialne
- postać o późnym początku
- dystrofinopatie (dystrofia mięśniowa Duchenne’a lub Beckera)
- dystrofie obręczowo-kończynowe
- zapalenie wielomięśniowe i inne miopatie autoimmunologiczne (w tym paranowotworowe)
- inne miopatie nabyte
- glikogenoza typu V (choroba McArdle’a)
- glikogenoza typu VI
Choroba Pompego – poradnictwo genetyczne
Choroba Pompego jest dziedziczona autosomalnie recesywnie. Oznacza to, że rodzice chorego dziecka ponoszą 25% ryzyko powtórzenia się tej samej choroby u kolejnych dzieci. Ryzyko to dotyczy każdej ciąży i nie zależy od płci dziecka. Przebieg kliniczny choroby będzie podobny u wszystkich chorych dzieci tych samych rodziców (tzn. jeżeli u jednego dziecka wystąpiła ciężka postać o wczesnym początku, kolejne chore dziecko również będzie miało taką postać choroby).
Jeżeli znane są mutacje patogenne u chorego dziecka, w kolejnej ciąży można rozważyć przeprowadzenie diagnostyki prenatalnej w kierunku choroby Pompego (co może być istotne dla szybkiego rozpoczęcia leczenia po urodzeniu).
Ryzyko wystąpienia choroby Pompego u dziecka osoby chorej jest niewielkie, ale każde dziecko takiej osoby będzie nosicielem mutacji patogennej w jednym allelu. W tej sytuacji choroba u dziecka mogłaby wystąpić tylko w przypadku, gdyby drugi z rodziców (tzn. partner lub partnerka chorej osoby) był nosicielem mutacji patogennej tego samego genu. Biorąc pod uwagę rzadkie występowanie choroby Pompego, taki stan nosicielstwa u partnera/partnerki jest mało prawdopodobny, ale nie można go wykluczyć bez wykonania badań genetycznych. Należy również pamiętać, że prawdopodobieństwo nosicielstwa mutacji może być znacznie większe, jeżeli partner/partnerka wykazuje pokrewieństwo z pacjentem. W każdym przypadku rozpoznania choroby Pompego wskazana jest konsultacja rodziny w poradni genetycznej.
Choroba Pompego – leczenie
Postępowanie terapeutyczne obejmuje leczenie przyczynowe za pomocą enzymatycznej terapii zastępczej oraz leczenie objawowe poszczególnych zaburzeń, np. niewydolności serca lub niewydolności oddechowej.
Enzymatyczna terapia zastępcza w chorobie Pompego
Leczenie przyczynowe choroby pompego polega na przewlekłej substytucji rekombinowanego analogu naturalnego enzymu. Lek podaje się w powolnym wlewie dożylnym co dwa tygodnie w dawce 20 mg/kg. W Polsce terapia jest w całości refundowana, zarówno u dzieci, jak i dorosłych bez ograniczenia wieku, w ramach programu leczenia chorób ultrarzadkich. Szpital, w którym będzie prowadzone leczenie, musi złożyć odpowiedni wniosek do Zespołu Koordynacyjnego ds. Chorób Ultrarzadkich. Warunkiem kwalifikacji pacjenta do leczenia jest potwierdzenie rozpoznania zarówno za pomocą testów enzymatycznych, jak i genetycznych. Zgodnie z wymogami programu terapeutycznego przed rozpoczęciem leczenia i następnie co 6 miesięcy należy przeprowadzać dokładną ocenę stanu klinicznego pacjenta, m.in. z uwzględnieniem echokardiografii, spirometrii oraz testów sprawności ruchowej. W razie nieskuteczności leczenia zgoda na refundację terapii może zostać wstrzymana. Ze względu na ryzyko reakcji nadwrażliwości, w tym wstrząsu anafilaktycznego, oraz ogólnie zwiększone ryzyko powikłań z powodu niewydolności oddechowej i/lub niewydolności serca leczenie należy prowadzić w szpitalu dysponującym odpowiednim oddziałem intensywnej opieki medycznej.
Ryzyko reakcji nadwrażliwości jest większe u niemowląt całkowicie pozbawionych naturalnego enzymu, u których wynik testu western blot na obecność alfa-glukozydazy w fibroblastach skóry jest ujemny. Większe jest również u nich ryzyko powstania przeciwciał znacząco hamujących działanie egzogennego enzymu. Z tego powodu w czasie enzymatycznej terapii zastępczej u tych dzieci proponuje się równoczesne stosowanie leków immunomodulujących, najczęściej rytuksymabu w połączeniu z innym lekiem immunosupresyjnym.
Wyniki dotychczasowych badań klinicznych wskazują, że u niemowląt i młodszych dzieci (<3,5 lat) enzymatyczna terapia zastępcza wydłuża przeżycie i okres bez konieczności stosowania wentylacji mechanicznej oraz zmniejsza masę serca i znacząco poprawia rozwój ruchowy. Skuteczność leczenia jest tym większa, im wcześniej zostanie ono rozpoczęte, najlepiej w pierwszych 2 tygodniach życia. W krajach, w których nie prowadzi się badań przesiewowych, tak wczesne rozpoznanie jest mało prawdopodobne. Możliwe jest jednak u kolejnych dzieci tych samych rodziców w przypadku wcześniejszego rozpoznania choroby Pompego u starszego potomstwa.
U pacjentów z postacią o późnym początku leczenie poprawia sprawność ruchową i zatrzymuje progresję niewydolności oddechowej. Poszczególne typy mięśni mogą być w różnym stopniu podatne na leczenie. Dostępne obecnie dane naukowe świadczą zgodnie, że większość leczonych odnosi istotne korzyści kliniczne.
Multimedia i nagrania
- Jaka jest skuteczność enzymatycznej terapii zastępczej u chorych na chorobę Pompego? – Link do artykułu
- Algorytm diagnostyki pacjentów z objawami dystrofii obręczowo-kończynowej z uwzględnieniem choroby Pompego – Link do artykułu
- Jakie cechy kliniczne mogą przemawiać za rozpoznaniem choroby Pompego? – Link do artykułu
- Czym wyróżnia się choroba Pompego wśród innych miopatii o fenotypie dystrofii obręczowo-kończynowej? – Link do artykułu
- Jakie jest znaczenie przesiewowych testów suchej kropli krwi w rozpoznawaniu choroby Pompego? – Link do artykułu
- Artykuł w języku angielskim o chorobie Pompego na Raredeseases.com – Link do artykułu
Choroba Pompego – infografika
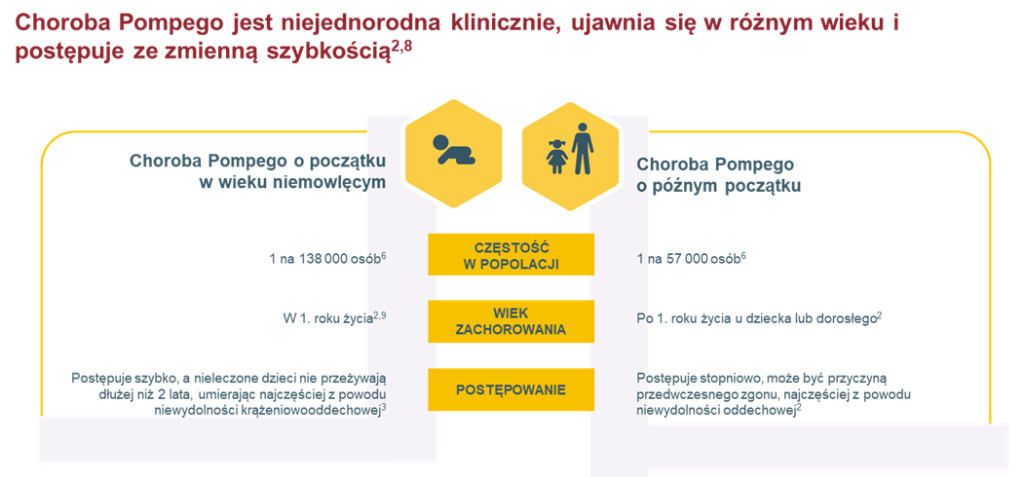
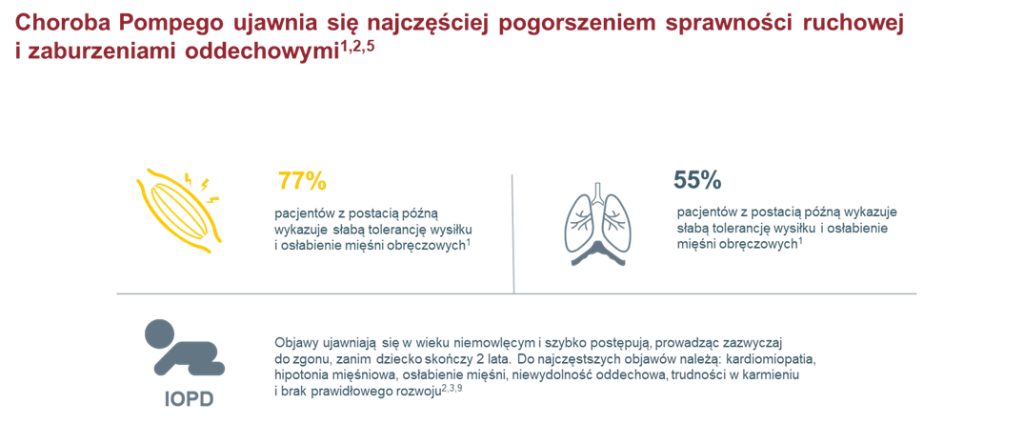

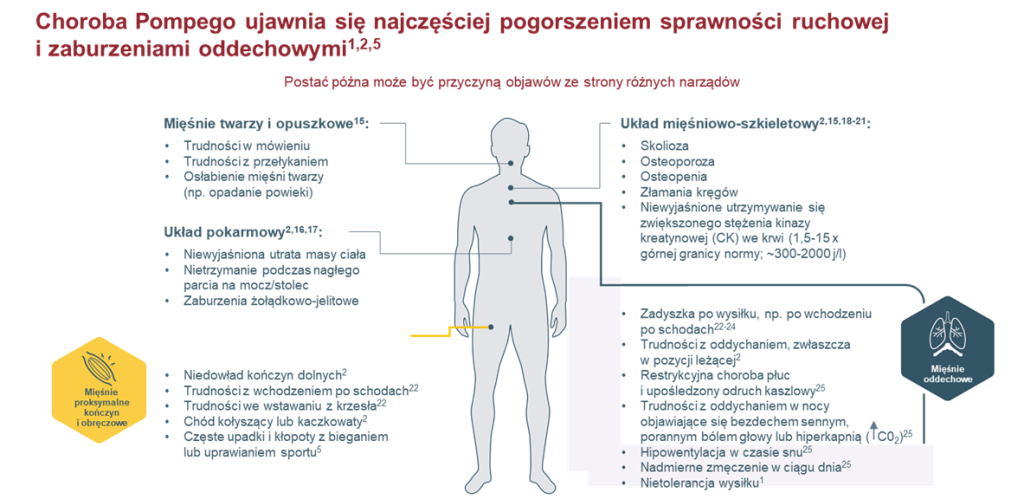
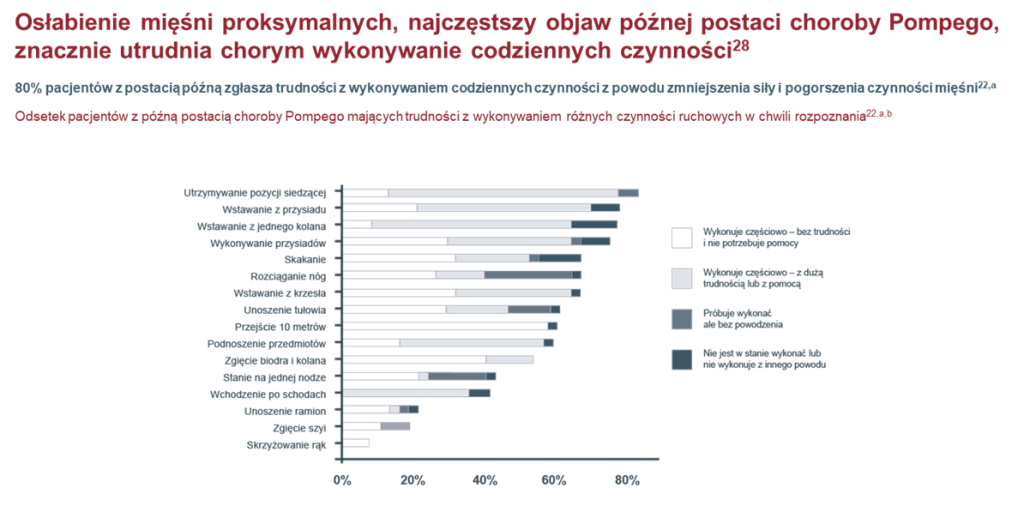

Co to jest M6P?
M6P (6-fosforan mannozy) jest częścią cząsteczki, która wiąże się z określonymi komórkami mięśniowymi receptory i pomaga enzymatycznej terapii zastępczej dotrzeć do komórek mięśniowych.

Czym jest choroba Pompego? Jak działa (awalglukozydaza alfa)?
Awalglukozydaza alfa ma ~15x WIĘCEJ M6P niż obecny standard opieki, alglukozydaza alfa (Lumizyme) i został stworzony w celu poprawy wychwytu enzymu przez komórki mięśniowe.
Choroba Pompego to zwyrodnieniowa choroba mięśni spowodowana niedoborem enzymu GAA.
W normalnej komórce mięśniowej enzym GAA rozkłada glikogen, rodzaj cukru, dzięki czemu komórka może go wykorzystać jako źródło energii. Dzieje się to w obszarze zwanym lizosomem. Aby się tam dostać, GAA potrzebuje pomocy M6P, która jest częścią cząsteczki.
Osoby z chorobą Pompego mają bardzo mało GAA lub nie mają go wcale, dlatego organizm nie może rozkładać glikogenu w mięśniach tak, jak powinien. Glikogen gromadzi się, powodując ciągłe uszkodzenie mięśni.
(awalglukozydaza alfa) to enzymatyczna terapia zastępcza wzbogacona w M6P stosowana w leczeniu choroby Pompego o późnym początku (LOPD). Pomaga zastąpić enzym GAA osobom, których organizmy nie produkują wystarczającej ilości. (awalglukozydaza alfa) został naukowo zaprojektowany tak, aby był skutecznie wchłaniany przez mięśnie.
Wwalglukozydaza alfa vs Alglukozydaza alfa
W badaniu kluczowym (badanie COMET) preparat (awalglukozydaza alfa) wykazał znaczną poprawę oddychania i chodzenia w porównaniu z alglukozydazą alfa.
Wszyscy (100 pacjentów w wieku 16-78 lat z LOPD) w badaniu mieli późną postać choroby Pompego, ale żaden z nich nie był wcześniej leczony. Niektórym podano (awalglukozydaza alfa), a innym alglukozydazę alfa. Wszystkie 100 osób było leczonych w badaniu przez 49 tygodni.
Użyto min. 2 testów do zmierzenia, jak nowy lek wpływa na chorobę i jak ludzie radzili sobie w badaniu, to testy:
WYMUSZONA POJEMNOŚĆ ŻYCIOWA (FVC) mierzy, ile powietrza możesz wydmuchać podczas głębokiego oddechu
6-MINUTOWY TEST WALK (6MWT) mierzy odległość, którą możesz przejść w tym przedziale czasowym
Testy przeprowadzono na początku i ponownie na końcu badania.
Po około 1 roku (49 tygodni) ludzie leczeni (awalglukozydaza alfa) byli w stanie:
Osoby, które przyjmowały alglukozydazę alfa, poprawiły oddychanie średnio o 0,5 punktu procentowego.
Osoby przyjmujące (awalglukozydaza alfa) szły średnio o 30 metrów dalej niż osoby przyjmujące alglukozydazę alfa. Tego pomiaru nie testowano w celu określenia statystycznej wyższości produktu (awalglukozydaza alfa) nad alglukozydazą alfa.
Osoby przyjmujące (awalglukozydaza alfa) były w stanie lepiej oddychać i chodzić dalej po około 1 roku (49 tygodniach) w porównaniu z początkiem. W rozszerzonym badaniu otwartym*, niezależnie od tego, czy pacjenci rozpoczęli stosowanie alglukozydazy alfa, czy przestawili się na (awalglukozydaza alfa) z alglukozydazy alfa po ~1 roku, byli w stanie utrzymać stabilność przez ~3 lata (145 tygodni); i nie zaobserwowano żadnych nowych obaw dotyczących bezpieczeństwa.
Po pierwszym ~1 roku (49 tygodni) badania 44 osoby stosujące alglukozydazę alfa przeszły na (awalglukozydaza alfa). Tymczasem 51 osób, które już przyjmowały (awalglukozydaza alfa), nadal go przyjmowało. Po około 3 latach (145 tygodni) otwartego przedłużenia badania większość osób przyjmujących (awalglukozydaza alfa) była w stanie utrzymać zdolność oddychania i chodzenia.
W badaniu klinicznym (awalglukozydaza alfa) wykazał podobne bezpieczeństwo i ryzyko jak alglukozydaza alfa. W trakcie 49-tygodniowego badania osoby przyjmujące (awalglukozydaza alfa) miały mniej poważnych działań niepożądanych niż osoby przyjmujące alglukozydazę alfa.† Nikt nie musiał przerywać przyjmowania (awalglukozydaza alfa) z powodu działań niepożądanych, natomiast 4 osoby musiały przerwać przyjmowanie alglukozydazy alfa.
CZĘSTE DZIAŁANIA NIEPOŻĄDANE (awalglukozydaza alfa) to:
- ból głowy, zmęczenie, zawroty głowy, mdłości, biegunka, wymioty, ból stawu, ból w mięśniach, swędzący, duszność, wysypka, wrażenie „szpilek i igieł”, pokrzywka.
Są to najczęstsze działania niepożądane, ale są też inne możliwe działania niepożądane. Zawsze należy poinformować lekarza o wszelkich zmianach w samopoczuciu, nawet jeśli nie jest to jedno z wymienionych działań niepożądanych. Działania niepożądane można również zgłaszać pod adresem:
Profil bezpieczeństwa produktu (awalglukozydaza alfa) został wykazany na podstawie prawie 7-letnich danych klinicznych z 4 badań klinicznych, które obejmowały osoby, które przeszły z alglukozydazy alfa.
†Badanie Comet nie zostało zaprojektowane w celu wykazania statystycznie istotnej różnicy w częstości występowania działań niepożądanych między produktem (awalglukozydaza alfa) a alglukozydazą alfa.
Piśmiennictwo, przypisy i źródła
- Schüller A, i wsp. Am J Med Genet C Semin Med Genet. 2012;160C(1):80-88. doi:10.1002/ajmg.c.31322. 2. Kishnani PS, i wsp.; ACMG Work Group on Management of Pompe Disease. Genet Med. 2006;8(5):267-288. doi:10.1097/01.gim.0000218152.87434.f3. 3. Kishnani PS, i wsp. J Pediatr. 2004;144(5 suppl):S35-S43. 5. van Capelle CI, i wsp. Orphanet J Rare Dis. 2016;11(1):65. doi:10.1186/s13023-016-0442-y. 9. Kishnani PS i wsp.; Infantile-Onset Pompe Disease Natural History Study Group. J Pediatr. 2006;148(5):671-676. doi:10.1016/j. jpeds.2005.11.033.
- Hirschhorn R i wsp. W: Scriver CR i wsp., eds. The Metabolic Bases of Inherited Disease. wyd. 8 New York, NY: McGraw-Hill; 2001:3389-3420. 12. Al Jasmi F i wsp.; the MENA Pompe Working Group. BMC Neurology. 2015;15:205. doi:10.1186/ s12883-015-0412-3. 13. Thurberg BL i wsp. Lab Invest. 2006;86(12):1208-1220. 14. Raben N i wsp. Mol Genet Metab. 2010;101(4):324-331. doi:10.1016/j.ymgme.2010.08.001. 15. Chan J i wsp. Mol Genet Metab. 2017;120(3):163-172. doi:10.1016/j.ymgme.2016.12.004. 16. Karabul N i wsp. JIMD Rep. 2014;17:53-61. doi:10.1007/8904_2014_334. 17. Bernstein DL i wsp. Mol Genet Metab. 2010;101(2-3):130-133. doi:10.1016/j.ymgme.2010.06.003. 18. Toscano A i wsp. Acta Myol. 2013;32(2): 78-81. 19. Preisler N i wsp. Mol Genet Metab. 2013;110(3):287-289. doi:10.1016/j.ymgme.2013.08.005. 20. Manganelli F i wsp. Acta Myol. 2013;32(2):82-84. 21. Moghadam-Kia S i wsp. Cleve Clin J Med. 2016;83(1):37-42. doi:10.3949/ccjm.83a.14120. 22. Rigter T i wsp. Mol Genet Metab. 2012;107(3):448-455. doi:0.1016/j.ymgme.2012.09.017. 23. Mellies U i wsp. Respir Med. 2009;103(4):477-484. doi:10.1016/j.rmed.2008.12.009. 24. Boentert M i wsp. Int J Mol Sci. 2016;17(10):1735. doi:10.3390/ijms17101735. 25. Fuller DD i wsp. Respir Physiol Neurobiol. 2013;189(2):241- 249. doi:10.1016/j.resp.2013.06.007. 26. Hagemans ML i wsp. Neurology. 2005;64(12):2139-2141. 28. Wokke JHJ i wsp. Muscle Nerve. 2008;38(4):1236-1245. doi:10.1002/ mus.21025. 29. Winkel LPF i wsp. J Neurol. 2005;252(80):875- 884. doi:10.1007/s00415-005-0922-9. 30. Schoser B i wsp. J Neurol. 2017;264(4):621-630. doi:10.1007/s00415-016-8219-8. 31. Jaradeh S. Muscle disorders affecting oral and pharyngeal swallowing. Strona internetowa GI Motility Online. https://www.nature.com/gimo/contents/pt1/full/gimo35.html. Opublikowano 16 maja 2006 r. Dostęp: 23.06.2022 r. 32. Chaudhuri A i wsp. Lancet. 2004;363(9413):978-988. doi:10.1016/S0140-6736(04)15794-2. 33. Hereditary myopathy with early respiratory failure. [Dziedziczna miopatia z wczesną niewydolnością oddechową.] Strona internetowa Genetics Home Reference. https://ghr.nlm.nih.gov/condition/hereditary-myopathy-with-early-respiratoryfailure. Weryfikacja: wrzesień 2018 r. Dostęp 28 lipca 2020 r. 34. Barohn RJ i wsp. Neurol Clin. 2014;32(3):569-vii. doi:10.1016/j.ncl.2014.04.008. 35. Féasson L i wsp. Ann Readapt Med Phys. 2006;49(6):289-300. doi:10.1016/j.annrmp.2006.04.015. 36. Gilchrist JM. Semin Respir Crit Care Med. 2002;23(3):191-200. doi:10.1055/s-2002-33027. 37. Ozawa E i wsp. Mol Cell Biochem. 1999;190:143-151. 38. Mah JK i wsp. Neuromuscul Disord. 2014;24(6):482-491. doi:10.1016/j.nmd.2014.03.008. 39. Barnabei MS i wsp. Compr Physiol. 2011;1(3):1353-1363. doi:10.1002/cphy.c100062. 40. Limb-girdle muscular dystrophy. [Dystrofia mięśni obręczy kończyny.] Strona internetowa Genetics Home Reference. https://ghr.nlm.nih.gov/condition/limb-girdlemuscular-dystrophy. Weryfikacja: wrzesień 2019 r. Dostęp 28 lipca 2020 r. 41. Pegoraro E, i wsp. W: Pagon RA i wsp., eds. GeneReviews. Seattle, WA: University of Washington, Seattle; 1993. https://www.ncbi.nlm.nih.gov/books/NBK1408/. Opublikowano 8 czerwca 2000 r. Zaktualizowano 30 sierpnia 2012 r. Dostęp 23.06.2022 r. 42. Siciliano G i wsp. Acta Myol. 2015;34(1):3-8. 43. Myasthenia gravis. [Miastenia] Strona internetowa Genetics Home Reference. https:// ghr.nlm.nih.gov/condition/myastheniagravis. Weryfikacja: czerwiec 2016 r. Dostęp 29 lipca 2020 r. 44. Myasthenia gravis: what is it? [Miastenia – co to jest?] Strona internetowa Harvard Health Publishing. https://www.health.harvard.edu/a_to_z/myasthenia-gravis-a-to-z. Opublikowano w grudniu 2018 r. Dostęp 23.06.2022 r. 45. Smoyer-Tomic KE i wsp. BMC Musculoskeletal Disord. 2012;13:103. doi:10.1186/1471-2474-13-103. 46. Strona informacyjna dotycząca miopatii zapalnych. Strona internetowa National Institute of Neurological Disorders and Stroke. https://www.ninds.nih.gov/Disorders/All-Disorders/Inflammatory-Myopathies-Information-Page. Opublikowano 27 lutego 2017 r. Dostęp 23.06.2022 r. 47. Gazeley DJ i wsp. Ther Adv Musculoskeletal Disord. 2011;3(6):315-324. doi:10.1177/1759720X11415306. 48. Polymyositis. [Zapalenie wielomięśniowe.] Strona internetowa Johns Hopkins Medicine. https://www.hopkinsmedicine.org/health/conditions-and-diseases/polymyositis. Opublikowano 27 lutego 2017 r. Dostęp 23.06.2022 r. 49. Mastaglia FL i wsp. Rheum Dis Clin North Am. 2002;28(4):723-741. doi:10.1016/s0889-857x(02)00021-2. 50. Wagner M i wsp. Neuromuscul Disord. 2013;23(1):89-92. doi:10.1016/j.nmd.2012.09.004. 51. Okumiya T i wsp. Mol Genet Metab. 2006;88(1):22-28. doi:10.1016/j.ymgme.2005.10.016. 52. Behjati S i wsp. Arch Dis Child Educ Pract Ed. 2013;98(6):236-238. doi:10.1136/archdischild-2013-304340. 53. Bodamer OA i wsp.; w imieniu grupy roboczej Pompe Disease Newborn Screening Working Group. Pediatrics. 2017;140(suppl 1):S4-S13. doi:10.1542/peds.2016-0280C. 54. Recommended uniform screening panel. [Zalecany jednolity panel badań przesiewowych.] Strona internetowa Health Resources and Services Administration. https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html. Weryfikacja: luty 2020 r. Dostęp 23.06.2022 r. 55. O’Callaghan C i wsp. Respirol Case Rep. 2016;4(5):e00178. doi:10.1002/rcr2.178. 56. Johns MW. Sleep. 1991;14(6):540-545. 57. Wood KL. Strona internetowa Merck Manual Professional Version. https://www.merckmanuals.com/professional/pulmonary-disorders/tests-of-pulmonary-function-pft/airflow,-lung-volumes,-and-flow-volumeloop. Zaktualizowano w kwietniu 2020 r. Dostęp 29 lipca 2020 r. 58. Messina Z i wsp. StatPearls. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih. gov/books/NBK551648/. Zaktualizowano 21 listopada 2019 r. Dostęp 29 lipca 2020 r. 59. Ortiz-Prado E i wsp. Am J Blood Res. 2019;9(1):1-14. 60. Barba-Romero MA, Barrot E i wsp. Rev Neurol. 2012;54(8):497-507. 61. Llerena JC Jr i wsp. Arq Neuropsiquiatr. 2016;74(2):166-176. doi:10.1590/0004-282X20150194. 62. van der Ploeg AT i wsp. Eur J Neurol. 2017;24(6):768-e31. doi:10.1111/ene.13285.